

GenBank is a user friendly reference database that allows the user to specificy different criteria
Javier Izquierdo, Ph.D.
Duration (01:03)
Author: Charles Xu, Ph.D.
After carefully designing an experiment, meticulous sample collection and sequencing, what is in some ways the most challenging step is bioinformatics analysis. Assuming you now have tens of thousands, if not millions, of DNA sequences (composed of A’s, T’s, C’s, and Gs), understanding how to approach the bioinformatics analysis is the crucial final phase.

The term "bioinformatics" is a portmanteau of "biology" and "informatics." It refers to the broad field that uses information technology, specifically computers, to analyze data derived from biological sources. Given the vast amounts of data generated in modern biological research, it has become impossible to interpret this information without the aid of computational tools. This is particularly true in high-throughput genetic sequence analysis, where bioinformatics plays an essential role. In the context of eDNA studies, bioinformatics can be broken down into three general steps: 1) demultiplexing, 2) creating consensus sequences, and 3) taxonomic identification.
Before going deeper into each step, it is important to note that prior knowledge of command-line computing is almost essential. While some software is available in more user-friendly formats, such as web platforms (e.g., DNA Subway or Galaxy) or R packages, most tools require command-line proficiency. If you are unfamiliar with or need a refresher on command-line computing, this resource provides a helpful introduction: [Learn Metabarcoding]
For those new to bioinformatics, we recommend reading through this page first to understand the general steps of bioinformatic data processing. Further down the page, you will find a list of bioinformatics platforms and software packages, including tools for processing data at each step of the bioinformatics workflow, as well as pipelines that offer “all-in-one” data processing solutions. To skip the basics, you may go directly to the section titled “Bioinformatics Software Options and Video Tutorials,” which also includes several video tutorials for reference.
Demultiplexing refers to the process of ensuring that each DNA sequence obtained from a sequencing platform is correctly matched with its corresponding sample label (Petit-Marty et al., 2023). Unless sequencing is conducted at extremely high depths where each sample occupies an entire flow cell, samples will typically undergo multiplexing. This involves artificially ligating or attaching a unique short DNA sequence (e.g., an index, barcode, or sequence tag) to the end of all nucleotide strands within a given sample. This allows DNA from different samples to be combined in a single sequencing run.
Before pooling, it is essential to normalize DNA concentrations across all samples based on the required number of sequences per sample. Despite the mixing, the unique tags ensure that the sequences can later be traced back to their original samples. Depending on the setup, this tagging process may be performed by you before submitting the samples, or the sequencing center may handle it as part of their services. If the center performs the multiplexing, they will typically also handle the demultiplexing. However, if you multiplexed your samples, be prepared to manage the demultiplexing process as well.
Consensus sequences are composite DNA sequences created from multiple individual sequences of the same region that have been aligned, typically resulting in what are known as Operational Taxonomic Units (OTUs). OTUs serve as summaries of groups of similar sequences, and various approaches exist for assigning them (Deiner et al., 2017; Liu et al., 2020). Consensus sequences are generated because they provide a consistent, representative sequence for each OTU, simplifying subsequent analyses. It is important to note that repeatedly analyzing very similar or identical sequences from the same sample will not yield new information and will only consume unnecessary computational resources.
Before generating consensus sequences, demultiplexed sequences must first be trimmed to remove several elements:
Once the sequences are trimmed, paired-end sequences—those originating from the same DNA fragment but from opposite strands (forward 5' → 3' and reverse 3' → 5')—can be merged using a minimum overlap sequence threshold. These merged sequences are then aligned to form consensus sequences, with ambiguity codes from the International Union of Pure and Applied Chemistry (IUPAC) (e.g., W = A or T, K = G or T, N = A, C, G, or T) used at positions of genetic variation. The similarity threshold for aligning consensus sequences typically falls around 97-98%, though this depends on the study’s objectives.
If your goal is to determine species presence from eDNA samples, it is important that each consensus sequence approximates individual species or higher taxonomic levels, such as genus or family. In this case, a consensus sequence for a particular taxon should be consistent across multiple samples, reliably defining each OTU.
However, if the focus is on population genetic diversity, a lower similarity threshold may be used to capture intraspecific variation. An alternative approach is the use of Amplicon Sequence Variants (ASVs), which are real biological sequences inferred by modeling amplification and sequencing errors. ASVs offer fine-resolution, distinguishing sequences by even a single nucleotide difference (Callahan et al., 2017). Studies have demonstrated that ASVs can be more sensitive in biodiversity surveys, particularly for detecting rare taxa (Porter & Hajibabaei, 2020).
The final step in the bioinformatic processing of metabarcoding sequence data is taxonomic identification, or taxonomic assignment. Whether you opt for OTUs or ASVs, the ultimate goal is to determine the taxonomic identity of your sequences, allowing you to answer the core question of your study: What organisms are present in my eDNA samples?
Generally, taxonomic assignment relies on the use of a reference sequence database. A list of reference databases is shown below. If a suitable reference database is unavailable, one must be created. These databases contain curated DNA sequences with verified taxonomic identities, allowing you to compare and match them with your eDNA sequences. However, reference databases vary significantly in size and applicability (Xiong et al., 2022).
It is important to recognize that species identification will not be possible if a taxonomically verified DNA barcode for that species does not exist. Larger databases offer broader coverage, but their size can make them difficult to manage and may increase the likelihood of spurious matches. In contrast, smaller, curated databases may focus on specific taxonomic groups, habitats, or geographically localized taxa. These can improve accuracy and efficiency but may introduce bias due to the initial assumptions made in selecting the reference sequences.
Before using reference sequence databases for taxonomic assignment, ensure that portions of sequences not targeted by your primers—such as the primer-binding sites themselves—are removed, as these do not reflect natural variation. The level of taxonomic resolution you achieve can vary widely, from broad categories (e.g., kingdom-level) to highly specific identifications (e.g., species or individual organism-level). The accuracy of taxonomic assignment depends on several factors, including the size and genetic variability of the target amplicon, the representation and specificity within your reference database, and the particular software, algorithm, or approach used for taxonomic assignment.
If possible, consider supplementing pre-existing databases by generating reference sequences from the tissues of taxonomically identified organisms relevant to your study. These supplemental sequences can enhance detection accuracy, particularly when the species or populations are the same as those targeted in your eDNA or metabarcoding study. This approach helps bridge gaps in existing databases and improves the reliability of species identification in your analyses
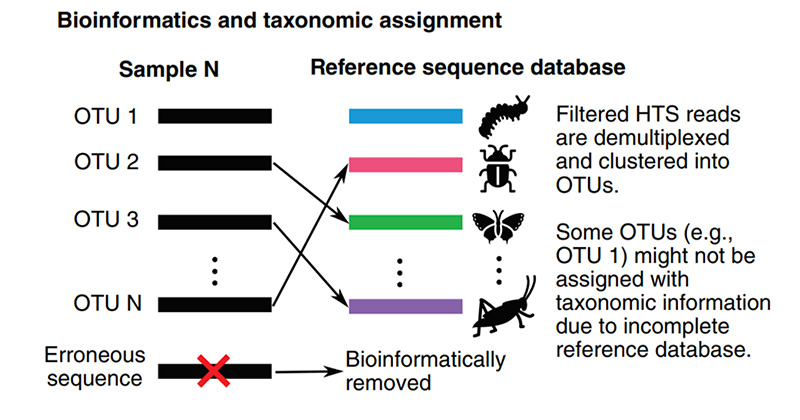
HTS = high-throughput sequencing. Adapted from Liu et al. 2020. (Image copyright policy: https://resjournals.onlinelibrary.wiley.com/hub/journal/13652311/homepage/forauthors.html)
The following are reference databases that you can use to determine the taxonomic identity of your sample:
While bioinformatic analysis involves numerous decision points, one of the advantages is that you can revisit previous steps, adjust parameters, and try alternative approaches, provided you have kept the intermediate files. At a minimum, you can always return to your original data, which underscores the importance of data storage and management.
Your original data must be stored securely and backed up multiple times. Even with advanced computing infrastructure, unforeseen issues can occur, making data redundancy critical. Intermediate files are useful to retain, as they reduce the need to rerun computationally intensive steps. However, these files can quickly grow in size, consuming valuable storage space. It is essential to strike a balance between storing intermediate files and managing storage efficiently based on the time it takes to regenerate them.
Additionally, FAIR data management principles—Findable, Accessible, Interoperable, and Reusable—should be prioritized. eDNA metabarcoding data often have value beyond the immediate study and may be utilized in future meta-analyses (Shea et al., 2023; Wilkinson et al., 2016). Implementing these strategies ensures that your data remain organized, accessible, and valuable for future research
Note that full or partial integration of software is needed for multiple processing steps and dependencies may need to be installed separately.
If the quality of your raw sequences is too low for successful read pairing, even with a low minimum overlap, you can attempt to process each single read individually. While this means you will lose the benefits of paired reads—such as confirmation of the overlapping sequence and longer contigs—you will still have some usable data. However, it is important to remember the computing principle: "garbage in, garbage out." Poor-quality input data will likely result in poor-quality output. If, after the initial quality check of your DNA extractions, you find that your sequences are unusable, you have two general options:
It is important to note that sequencing centers often have higher quality standards than necessary to ensure excellent data for their clients. Therefore, maintaining open and transparent communication with the sequencing center's technical support team is crucial. Their expertise can guide you through these challenges, and as a paying customer, you should take full advantage of their advice.
The answer to this question depends on several factors. Ideally, you will want to set parameters that exclude unreliable sequences while maximizing the retention of samples in your dataset. For example, if your low-quality samples are relatively evenly distributed across your treatments, you may be able to exclude those samples and still conduct valid comparisons across treatments. The exact parameters will vary depending on the sequencing platform, sequencing depth, quality score distribution, and bioinformatics pipeline used
ASVs (Amplicon Sequence Variants) are generally recommended over OTUs (Operational Taxonomic Units) because each ASV represents a biologically meaningful sequence, and there are demonstrable benefits to ASV analysis compared to OTU analysis (Callahan et al., 2017). However, if you prefer OTUs, they are still widely accepted in the literature and often provide similar data interpretations. It may be helpful to review studies that explicitly compare the two approaches before deciding which analysis to use (Chiarello et al., 2022; García-López et al., 2021; Jeske & Gallert, 2022; Joos et al., 2020; Prodan et al., 2020).
As with many bioinformatic questions, there is no simple answer. The best method is ultimately the one that allows you to achieve your scientific objectives within the time available. For example, if you have little or no bioinformatics experience, it may be best to start with a user-friendly pipeline that offers a graphical user interface (GUI). Additionally, choosing a well-maintained pipeline with extensive support is advantageous, as you can benefit from an active user community and expert advice. For instance, QIIME2 has a very active user forum, making it a solid choice for beginners.
Due to discrepancies between different taxonomic assignment approaches, it is also considered good practice to use a combination of software platforms to verify that they yield consistent results. This will help ensure the robustness and reliability of your analysis
If you are interested in specific organisms, it is important to verify that their reference sequences are included in your database. One way to test the reliability of your reference database is to use mock communities—groups of organisms for which the taxonomic identities are already known. Mock communities can be generated de novo from DNA samples of taxonomically confirmed organisms or in silico using sequence data from your reference database. In addition to validating the reference database, mock communities are valuable for benchmarking other aspects of your bioinformatic workflow, such as filtration parameters and taxonomic assignment methods
Open access:
Not open access but very useful nonetheless:
Abdala Asbun, A., Besseling, M. A., Balzano, S., Van Bleijswijk, J. D. L., Witte, H. J., Villanueva, L., & Engelmann, J. C. (2020). Cascabel: A Scalable and Versatile Amplicon Sequence Data Analysis Pipeline Delivering Reproducible and Documented Results. Frontiers in Genetics, 11, 489357. https://doi.org/10.3389/fgene.2020.489357
Albanese, D., Fontana, P., De Filippo, C., Cavalieri, D., & Donati, C. (2015). MICCA: A complete and accurate software for taxonomic profiling of metagenomic data. Scientific Reports, 5(1), 9743. https://doi.org/10.1038/srep09743
Allard, G., Ryan, F. J., Jeffery, I. B., & Claesson, M. J. (2015). SPINGO: A rapid species-classifier for microbial amplicon sequences. BMC Bioinformatics, 16(1), 324. https://doi.org/10.1186/s12859-015-0747-1
Anslan, S., Bahram, M., Hiiesalu, I., & Tedersoo, L. (2017). PipeCraft: Flexible open‐source toolkit for bioinformatics analysis of custom high‐throughput amplicon sequencing data. Molecular Ecology Resources, 17(6). https://doi.org/10.1111/1755-0998.12692
Ansorge, R., Birolo, G., James, S. A., & Telatin, A. (2021). Dadaist2: A Toolkit to Automate and Simplify Statistical Analysis and Plotting of Metabarcoding Experiments. International Journal of Molecular Sciences, 22(10), 5309. https://doi.org/10.3390/ijms22105309
Barbera, P., Kozlov, A. M., Czech, L., Morel, B., Darriba, D., Flouri, T., & Stamatakis, A. (2019). EPA-ng: Massively Parallel Evolutionary Placement of Genetic Sequences. Systematic Biology, 68(2), 365–369. https://doi.org/10.1093/sysbio/syy054
Bolyen, E., Rideout, J. R., Dillon, M. R., Bokulich, N. A., Abnet, C. C., Al-Ghalith, G. A., Alexander, H., Alm, E. J., Arumugam, M., Asnicar, F., Bai, Y., Bisanz, J. E., Bittinger, K., Brejnrod, A., Brislawn, C. J., Brown, C. T., Callahan, B. J., Caraballo-Rodríguez, A. M., Chase, J., … Caporaso, J. G. (2019). Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nature Biotechnology, 37(8), 852–857. https://doi.org/10.1038/s41587-019-0209-9
Bouskill, N. J., Lim, H. C., Borglin, S., Salve, R., Wood, T. E., Silver, W. L., & Brodie, E. L. (2013). Pre-exposure to drought increases the resistance of tropical forest soil bacterial communities to extended drought. The ISME Journal, 7(2), 384–394. https://doi.org/10.1038/ismej.2012.113
Buchner, D., Macher, T.-H., & Leese, F. (2022). APSCALE: Advanced pipeline for simple yet comprehensive analyses of DNA metabarcoding data. Bioinformatics, 38(20), 4817–4819. https://doi.org/10.1093/bioinformatics/btac588
Cai, Y., Zheng, W., Yao, J., Yang, Y., Mai, V., Mao, Q., & Sun, Y. (2017). ESPRIT-Forest: Parallel clustering of massive amplicon sequence data in subquadratic time. PLOS Computational Biology, 13(4), e1005518. https://doi.org/10.1371/journal.pcbi.1005518
Callahan, B. J., McMurdie, P. J., & Holmes, S. P. (2017). Exact sequence variants should replace operational taxonomic units in marker-gene data analysis. The ISME Journal, 11(12), 2639–2643. https://doi.org/10.1038/ismej.2017.119
Callahan, B. J., McMurdie, P. J., Rosen, M. J., Han, A. W., Johnson, A. J. A., & Holmes, S. P. (2016). DADA2: High-resolution sample inference from Illumina amplicon data. Nature Methods, 13(7), 581–583. https://doi.org/10.1038/nmeth.3869
Camacho, C., Coulouris, G., Avagyan, V., Ma, N., Papadopoulos, J., Bealer, K., & Madden, T. L. (2009). BLAST+: Architecture and applications. BMC Bioinformatics, 10(1), 421. https://doi.org/10.1186/1471-2105-10-421
Chiarello, M., McCauley, M., Villéger, S., & Jackson, C. R. (2022). Ranking the biases: The choice of OTUs vs. ASVs in 16S rRNA amplicon data analysis has stronger effects on diversity measures than rarefaction and OTU identity threshold. PLOS ONE, 17(2), e0264443. https://doi.org/10.1371/journal.pone.0264443
Deiner, K., Bik, H. M., Mächler, E., Seymour, M., Lacoursière-Roussel, A., Altermatt, F., Creer, S., Bista, I., Lodge, D. M., de Vere, N., Pfrender, M. E., & Bernatchez, L. (2017). Environmental DNA metabarcoding: Transforming how we survey animal and plant communities. Molecular Ecology, 26(21), 5872–5895. https://doi.org/10.1111/mec.14350
Djemiel, C., Dequiedt, S., Karimi, B., Cottin, A., Girier, T., El Djoudi, Y., Wincker, P., Lelièvre, M., Mondy, S., Chemidlin Prévost-Bouré, N., Maron, P.-A., Ranjard, L., & Terrat, S. (2020). BIOCOM-PIPE: A new user-friendly metabarcoding pipeline for the characterization of microbial diversity from 16S, 18S and 23S rRNA gene amplicons. BMC Bioinformatics, 21(1), 492. https://doi.org/10.1186/s12859-020-03829-3
Dufresne, Y., Lejzerowicz, F., Perret-Gentil, L. A., Pawlowski, J., & Cordier, T. (2019). SLIM: A flexible web application for the reproducible processing of environmental DNA metabarcoding data. BMC Bioinformatics, 20(1), 88. https://doi.org/10.1186/s12859-019-2663-2
Edgar, R. C. (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics, 26(19), 2460–2461. https://doi.org/10.1093/bioinformatics/btq461
Edgar, R. C. (2013). UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nature Methods, 10(10), 996–998. https://doi.org/10.1038/nmeth.2604
Escudié, F., Auer, L., Bernard, M., Mariadassou, M., Cauquil, L., Vidal, K., Maman, S., Hernandez-Raquet, G., Combes, S., & Pascal, G. (2018). FROGS: Find, Rapidly, OTUs with Galaxy Solution. Bioinformatics, 34(8), 1287–1294. https://doi.org/10.1093/bioinformatics/btx791
Ewels, P. A., Peltzer, A., Fillinger, S., Patel, H., Alneberg, J., Wilm, A., Garcia, M. U., Di Tommaso, P., & Nahnsen, S. (2020). The nf-core framework for community-curated bioinformatics pipelines. Nature Biotechnology, 38(3), 276–278. https://doi.org/10.1038/s41587-020-0439-x
Fu, L., Niu, B., Zhu, Z., Wu, S., & Li, W. (2012). CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics, 28(23), 3150–3152. https://doi.org/10.1093/bioinformatics/bts565
García-López, R., Cornejo-Granados, F., Lopez-Zavala, A. A., Cota-Huízar, A., Sotelo-Mundo, R. R., Gómez-Gil, B., & Ochoa-Leyva, A. (2021). OTUs and ASVs Produce Comparable Taxonomic and Diversity from Shrimp Microbiota 16S Profiles Using Tailored Abundance Filters. Genes, 12(4), 564. https://doi.org/10.3390/genes12040564
Ghodsi, M., Liu, B., & Pop, M. (2011). DNACLUST: Accurate and efficient clustering of phylogenetic marker genes. BMC Bioinformatics, 12(1), 271. https://doi.org/10.1186/1471-2105-12-271
Guillou, L., Bachar, D., Audic, S., Bass, D., Berney, C., Bittner, L., Boutte, C., Burgaud, G., De Vargas, C., Decelle, J., Del Campo, J., Dolan, J. R., Dunthorn, M., Edvardsen, B., Holzmann, M., Kooistra, W. H. C. F., Lara, E., Le Bescot, N., Logares, R., … Christen, R. (2012). The Protist Ribosomal Reference database (PR2): A catalog of unicellular eukaryote Small Sub-Unit rRNA sequences with curated taxonomy. Nucleic Acids Research, 41(D1), D597–D604. https://doi.org/10.1093/nar/gks1160
Gweon, H. S., Oliver, A., Taylor, J., Booth, T., Gibbs, M., Read, D. S., Griffiths, R. I., & Schonrogge, K. (2015). PIPITS: An automated pipeline for analyses of fungal internal transcribed spacer sequences from the I llumina sequencing platform. Methods in Ecology and Evolution, 6(8), 973–980. https://doi.org/10.1111/2041-210X.12399
Hao, X., Jiang, R., & Chen, T. (2011). Clustering 16S rRNA for OTU prediction: A method of unsupervised Bayesian clustering. Bioinformatics, 27(5), 611–618. https://doi.org/10.1093/bioinformatics/btq725
Hupfauf, S., Etemadi, M., Fernández-Delgado Juárez, M., Gómez-Brandón, M., Insam, H., & Podmirseg, S. M. (2020). CoMA – an intuitive and user-friendly pipeline for amplicon-sequencing data analysis. PLOS ONE, 15(12), e0243241. https://doi.org/10.1371/journal.pone.0243241
Huson, D. H., Beier, S., Flade, I., Górska, A., El-Hadidi, M., Mitra, S., Ruscheweyh, H.-J., & Tappu, R. (2016). MEGAN Community Edition—Interactive Exploration and Analysis of Large-Scale Microbiome Sequencing Data. PLOS Computational Biology, 12(6), e1004957. https://doi.org/10.1371/journal.pcbi.1004957
Jeske, J. T., & Gallert, C. (2022). Microbiome Analysis via OTU and ASV-Based Pipelines—A Comparative Interpretation of Ecological Data in WWTP Systems. Bioengineering, 9(4), 146. https://doi.org/10.3390/bioengineering9040146
Jones, M., Ghoorah, A., & Blaxter, M. (2011). jMOTU and Taxonerator: Turning DNA Barcode Sequences into Annotated Operational Taxonomic Units. PLoS ONE, 6(4), e19259. https://doi.org/10.1371/journal.pone.0019259
Joos, L., Beirinckx, S., Haegeman, A., Debode, J., Vandecasteele, B., Baeyen, S., Goormachtig, S., Clement, L., & De Tender, C. (2020). Daring to be differential: Metabarcoding analysis of soil and plant-related microbial communities using amplicon sequence variants and operational taxonomical units. BMC Genomics, 21(1), 733. https://doi.org/10.1186/s12864-020-07126-4
Kahlke, T., & Ralph, P. J. (2019). BASTA – Taxonomic classification of sequences and sequence bins using last common ancestor estimations. Methods in Ecology and Evolution, 10(1), 100–103. https://doi.org/10.1111/2041-210X.13095
Kallio, M. A., Tuimala, J. T., Hupponen, T., Klemelä, P., Gentile, M., Scheinin, I., Koski, M., Käki, J., & Korpelainen, E. I. (2011). Chipster: User-friendly analysis software for microarray and other high-throughput data. BMC Genomics, 12(1), 507. https://doi.org/10.1186/1471-2164-12-507
Leray, M., Ho, S.-L., Lin, I.-J., & Machida, R. J. (2018). MIDORI server: A webserver for taxonomic assignment of unknown metazoan mitochondrial-encoded sequences using a curated database. Bioinformatics, 34(21), 3753–3754. https://doi.org/10.1093/bioinformatics/bty454
Leray, M., Knowlton, N., & Machida, R. J. (2022). MIDORI2: A collection of quality controlled, preformatted, and regularly updated reference databases for taxonomic assignment of eukaryotic mitochondrial sequences. Environmental DNA, 4(4), 894–907. https://doi.org/10.1002/edn3.303
Li, R., Ratnasingham, S., Zarubiieva, I., Somervuo, P., & Taylor, G. W. (2024). PROTAX-GPU: A scalable probabilistic taxonomic classification system for DNA barcodes. Philosophical Transactions of the Royal Society B: Biological Sciences, 379(1904), 20230124. https://doi.org/10.1098/rstb.2023.0124
Liu, M., Clarke, L. J., Baker, S. C., Jordan, G. J., & Burridge, C. P. (2020). A practical guide to DNA metabarcoding for entomological ecologists. Ecological Entomology, 45(3), 373–385. https://doi.org/10.1111/een.12831
Loos, D., Zhang, L., Beemelmanns, C., Kurzai, O., & Panagiotou, G. (2021). DAnIEL: A User-Friendly Web Server for Fungal ITS Amplicon Sequencing Data. Frontiers in Microbiology, 12, 720513. https://doi.org/10.3389/fmicb.2021.720513
Lu, J., Rincon, N., Wood, D. E., Breitwieser, F. P., Pockrandt, C., Langmead, B., Salzberg, S. L., & Steinegger, M. (2022). Metagenome analysis using the Kraken software suite. Nature Protocols, 17(12), 2815–2839. https://doi.org/10.1038/s41596-022-00738-y
Lu, Y., Zhou, G., Ewald, J., Pang, Z., Shiri, T., & Xia, J. (2023). MicrobiomeAnalyst 2.0: Comprehensive statistical, functional and integrative analysis of microbiome data. Nucleic Acids Research, 51(W1), W310–W318. https://doi.org/10.1093/nar/gkad407
Mahé, F., Czech, L., Stamatakis, A., Quince, C., De Vargas, C., Dunthorn, M., & Rognes, T. (2021). Swarm v3: Towards tera-scale amplicon clustering. Bioinformatics, 38(1), 267–269. https://doi.org/10.1093/bioinformatics/btab493
Martin, M. (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.Journal, 17(1), 10. https://doi.org/10.14806/ej.17.1.200
Mathon, L., Valentini, A., Guérin, P., Normandeau, E., Noel, C., Lionnet, C., Boulanger, E., Thuiller, W., Bernatchez, L., Mouillot, D., Dejean, T., & Manel, S. (2021). Benchmarking bioinformatic tools for fast and accurate eDNA metabarcoding species identification. Molecular Ecology Resources, 21(7), 2565–2579. https://doi.org/10.1111/1755-0998.13430
Matsen, F. A., Kodner, R. B., & Armbrust, E. V. (2010). pplacer: Linear time maximum-likelihood and Bayesian phylogenetic placement of sequences onto a fixed reference tree. BMC Bioinformatics, 11(1), 538. https://doi.org/10.1186/1471-2105-11-538
McDonald, D., Jiang, Y., Balaban, M., Cantrell, K., Zhu, Q., Gonzalez, A., Morton, J. T., Nicolaou, G., Parks, D. H., Karst, S. M., Albertsen, M., Hugenholtz, P., DeSantis, T., Song, S. J., Bartko, A., Havulinna, A. S., Jousilahti, P., Cheng, S., Inouye, M., … Knight, R. (2024). Greengenes2 unifies microbial data in a single reference tree. Nature Biotechnology, 42(5), 715–718. https://doi.org/10.1038/s41587-023-01845-1
Mock, F., Kretschmer, F., Kriese, A., Böcker, S., & Marz, M. (2022). Taxonomic classification of DNA sequences beyond sequence similarity using deep neural networks. Proceedings of the National Academy of Sciences, 119(35), e2122636119. https://doi.org/10.1073/pnas.2122636119
Mousavi‐Derazmahalleh, M., Stott, A., Lines, R., Peverley, G., Nester, G., Simpson, T., Zawierta, M., De La Pierre, M., Bunce, M., & Christophersen, C. T. (2021). eDNAFlow, an automated, reproducible and scalable workflow for analysis of environmental DNA sequences exploiting Nextflow and Singularity. Molecular Ecology Resources, 21(5), 1697–1704. https://doi.org/10.1111/1755-0998.13356
Özkurt, E., Fritscher, J., Soranzo, N., Ng, D. Y. K., Davey, R. P., Bahram, M., & Hildebrand, F. (2022). LotuS2: An ultrafast and highly accurate tool for amplicon sequencing analysis. Microbiome, 10(1), 176. https://doi.org/10.1186/s40168-022-01365-1
Palmer, J. M., Jusino, M. A., Banik, M. T., & Lindner, D. L. (2018). Non-biological synthetic spike-in controls and the AMPtk software pipeline improve mycobiome data. PeerJ, 6, e4925. https://doi.org/10.7717/peerj.4925
Petit-Marty, N., Casas, L., & Saborido-Rey, F. (2023). State-of-the-art of data analyses in environmental DNA approaches towards its applicability to sustainable fisheries management. Frontiers in Marine Science, 10. https://doi.org/10.3389/fmars.2023.1061530
Porter, T. M., & Hajibabaei, M. (2020). Putting COI Metabarcoding in Context: The Utility of Exact Sequence Variants (ESVs) in Biodiversity Analysis. Frontiers in Ecology and Evolution, 8, 248. https://doi.org/10.3389/fevo.2020.00248
Porter, T. M., & Hajibabaei, M. (2022). MetaWorks: A flexible, scalable bioinformatic pipeline for high-throughput multi-marker biodiversity assessments. PLOS ONE, 17(9), e0274260. https://doi.org/10.1371/journal.pone.0274260
Prodan, A., Tremaroli, V., Brolin, H., Zwinderman, A. H., Nieuwdorp, M., & Levin, E. (2020). Comparing bioinformatic pipelines for microbial 16S rRNA amplicon sequencing. PLOS ONE, 15(1), e0227434. https://doi.org/10.1371/journal.pone.0227434
Quast, C., Pruesse, E., Yilmaz, P., Gerken, J., Schweer, T., Yarza, P., Peplies, J., & Glöckner, F. O. (2012). The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Research, 41(D1), D590–D596. https://doi.org/10.1093/nar/gks1219
Ratnasingham, S. (2019). mBRAVE: The Multiplex Barcode Research And Visualization Environment. Biodiversity Information Science and Standards, 3, e37986. https://doi.org/10.3897/biss.3.37986
Ratnasingham, S., & Hebert, P. D. N. (2007). bold: The Barcode of Life Data System (http://www.barcodinglife.org). Molecular Ecology Notes, 7(3), 355–364. https://doi.org/10.1111/j.1471-8286.2007.01678.x
Rognes, T., Flouri, T., Nichols, B., Quince, C., & Mahé, F. (2016). VSEARCH: A versatile open source tool for metagenomics. PeerJ, 4, e2584. https://doi.org/10.7717/peerj.2584
Russell, D. J., Way, S. F., Benson, A. K., & Sayood, K. (2010). A grammar-based distance metric enables fast and accurate clustering of large sets of 16S sequences. BMC Bioinformatics, 11(1), 601. https://doi.org/10.1186/1471-2105-11-601
Sayers, E. W., Bolton, E. E., Brister, J. R., Canese, K., Chan, J., Comeau, D. C., Connor, R., Funk, K., Kelly, C., Kim, S., Madej, T., Marchler-Bauer, A., Lanczycki, C., Lathrop, S., Lu, Z., Thibaud-Nissen, F., Murphy, T., Phan, L., Skripchenko, Y., … Sherry, S. T. (2022). Database resources of the national center for biotechnology information. Nucleic Acids Research, 50(D1), D20–D26. https://doi.org/10.1093/nar/gkab1112
Schloss, P. D. (2020). Reintroducing mothur: 10 Years Later. Applied and Environmental Microbiology, 86(2), e02343-19. https://doi.org/10.1128/AEM.02343-19
Shah, N., Molloy, E. K., Pop, M., & Warnow, T. (2021). TIPP2: Metagenomic taxonomic profiling using phylogenetic markers. Bioinformatics, 37(13), 1839–1845. https://doi.org/10.1093/bioinformatics/btab023
Shea, M. M., Kuppermann, J., Rogers, M. P., Smith, D. S., Edwards, P., & Boehm, A. B. (2023). Systematic review of marine environmental DNA metabarcoding studies: Toward best practices for data usability and accessibility. PeerJ, 11, e14993. https://doi.org/10.7717/peerj.14993
Somervuo, P., Koskela, S., Pennanen, J., Henrik Nilsson, R., & Ovaskainen, O. (2016). Unbiased probabilistic taxonomic classification for DNA barcoding. Bioinformatics, 32(19), 2920–2927. https://doi.org/10.1093/bioinformatics/btw346
Stark, M., Berger, S. A., Stamatakis, A., & Von Mering, C. (2010). MLTreeMap—Accurate Maximum Likelihood placement of environmental DNA sequences into taxonomic and functional reference phylogenies. BMC Genomics, 11(1), 461. https://doi.org/10.1186/1471-2164-11-461
Thompson, L. R., Anderson, S. R., Den Uyl, P. A., Patin, N. V., Lim, S. J., Sanderson, G., & Goodwin, K. D. (2022). Tourmaline: A containerized workflow for rapid and iterable amplicon sequence analysis using QIIME 2 and Snakemake. GigaScience, 11, giac066. https://doi.org/10.1093/gigascience/giac066
Vasar, M., Davison, J., Neuenkamp, L., Sepp, S., Young, J. P. W., Moora, M., & Öpik, M. (2021). User‐friendly bioinformatics pipeline gDAT (graphical downstream analysis tool) for analysing rDNA sequences. Molecular Ecology Resources, 21(4), 1380–1392. https://doi.org/10.1111/1755-0998.13340
Větrovský, T., Baldrian, P., & Morais, D. (2018). SEED 2: A user-friendly platform for amplicon high-throughput sequencing data analyses. Bioinformatics, 34(13), 2292–2294. https://doi.org/10.1093/bioinformatics/bty071
Wei, Z.-G., & Zhang, S.-W. (2017). DBH: A de Bruijn graph-based heuristic method for clustering large-scale 16S rRNA sequences into OTUs. Journal of Theoretical Biology, 425, 80–87. https://doi.org/10.1016/j.jtbi.2017.04.019
Wei, Z.-G., & Zhang, S.-W. (2019). DMSC: A Dynamic Multi-Seeds Method for Clustering 16S rRNA Sequences Into OTUs. Frontiers in Microbiology, 10, 428. https://doi.org/10.3389/fmicb.2019.00428
Weißbecker, C., Schnabel, B., & Heintz-Buschart, A. (2020). Dadasnake, a Snakemake implementation of DADA2 to process amplicon sequencing data for microbial ecology. GigaScience, 9(12), giaa135. https://doi.org/10.1093/gigascience/giaa135
Wilkinson, M. D., Dumontier, M., Aalbersberg, Ij. J., Appleton, G., Axton, M., Baak, A., Blomberg, N., Boiten, J.-W., Da Silva Santos, L. B., Bourne, P. E., Bouwman, J., Brookes, A. J., Clark, T., Crosas, M., Dillo, I., Dumon, O., Edmunds, S., Evelo, C. T., Finkers, R., … Mons, B. (2016). The FAIR Guiding Principles for scientific data management and stewardship. Scientific Data, 3(1), 160018. https://doi.org/10.1038/sdata.2016.18
Wood, D. E., Lu, J., & Langmead, B. (2019). Improved metagenomic analysis with Kraken 2. Genome Biology, 20(1), 257. https://doi.org/10.1186/s13059-019-1891-0
Wright, E., S. (2016). Using DECIPHER v2.0 to Analyze Big Biological Sequence Data in R. The R Journal, 8(1), 352. https://doi.org/10.32614/RJ-2016-025
Xiong, F., Shu, L., Zeng, H., Gan, X., He, S., & Peng, Z. (2022). Methodology for fish biodiversity monitoring with environmental DNA metabarcoding: The primers, databases and bioinformatic pipelines. Water Biology and Security, 1(1), 100007. https://doi.org/10.1016/j.watbs.2022.100007
Zafeiropoulos, H., Viet, H. Q., Vasileiadou, K., Potirakis, A., Arvanitidis, C., Topalis, P., Pavloudi, C., & Pafilis, E. (2020). PEMA: A flexible Pipeline for Environmental DNA Metabarcoding Analysis of the 16S/18S ribosomal RNA, ITS, and COI marker genes. GigaScience, 9(3), giaa022. https://doi.org/10.1093/gigascience/giaa022
Zheng, Q., Bartow-McKenney, C., Meisel, J. S., & Grice, E. A. (2018). HmmUFOtu: An HMM and phylogenetic placement based ultra-fast taxonomic assignment and OTU picking tool for microbiome amplicon sequencing studies. Genome Biology, 19(1), 82. https://doi.org/10.1186/s13059-018-1450-0
Zheng, Z., Kramer, S., & Schmidt, B. (2012). DySC: Software for greedy clustering of 16S rRNA reads. Bioinformatics, 28(16), 2182–2183. https://doi.org/10.1093/bioinformatics/bts355
Zito, A., Rigon, T., & Dunson, D. B. (2023). Inferring taxonomic placement from DNA barcoding aiding in discovery of new taxa. Methods in Ecology and Evolution, 14(2), 529–542. https://doi.org/10.1111/2041-210X.14009
Duration (01:03)

Duration (01:30)

Duration (00:55)
Duration (00:59)